生物信息学基础与算法
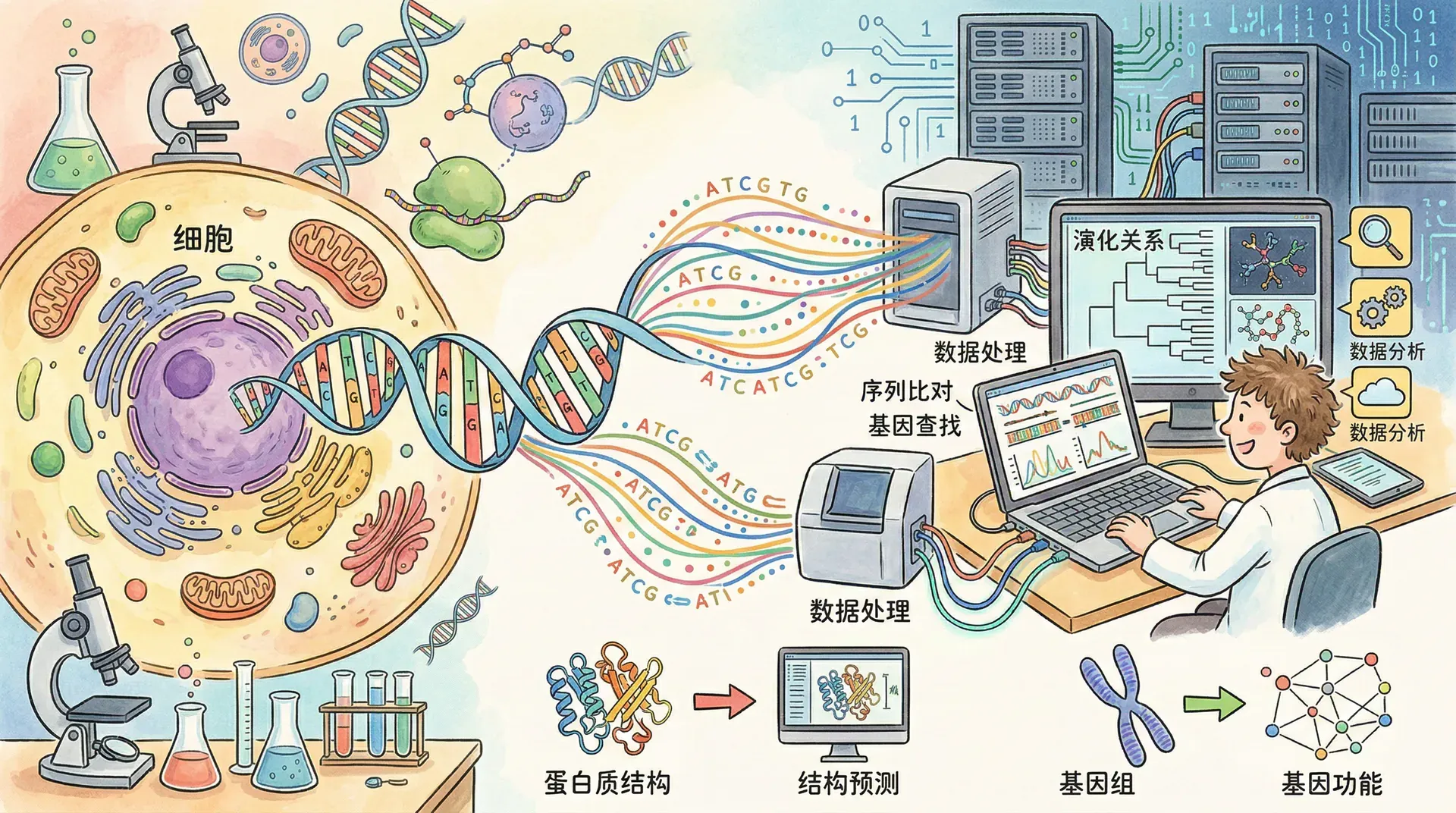
生物信息学是一门融合生物学、计算机科学、数学和统计学等多学科知识的交叉学科,旨在应对基因组测序等高通量技术带来的大数据挑战。随着DNA、RNA和蛋白质等分子的序列与结构信息被大规模获取,生物学数据呈现爆炸式增长,单个基因组测序项目就可产生TB甚至PB级的数据,这对数据的存储、管理和分析提出了巨大挑战。
为此,生物信息学侧重利用算法和软件工具管理与分析海量数据,并将复杂的生物学问题转化为计算模型,借助定量和系统的方法探索生命现象。通过这些计算方法,我们不仅能对基因组进行注释,预测基因和蛋白质功能,分析序列相似性及进化关系,还能研究生命系统的调控网络。此外,生物信息学在医学、农业、环境以及药物研发等领域也具有重要应用价值,包括疾病相关基因识别、新药靶点发现和作物基因改良等。
因此,生物信息学不仅仅是一门将生物学与信息技术相结合的新兴学科,更是一把开启生命科学新时代钥匙的工具,通过开发和应用各种计算方法,帮助科学家们从浩瀚的生物数据中挖掘和提取有价值的信息,推动生命科学研究的不断进步和创新。
生物信息学的研究内容与工具
生物信息学的核心任务是处理和分析各类生物学数据。这些数据包括DNA序列、RNA序列、蛋白质序列、蛋白质结构以及基因表达数据等。在中国,生物信息学的发展始于20世纪90年代,随着人类基因组计划的启动,中国科学家积极参与其中,承担了人类3号染色体短臂上约3000万个碱基对的测序任务。这一参与不仅提升了中国在基因组学领域的国际地位,也推动了国内生物信息学的快速发展。
生物信息学的研究内容可以分为几个主要方向。
生物信息学的发展离不开强大的计算工具和数据库。目前,国际上有许多公开的生物信息学数据库和工具供研究者免费使用,如NCBI的GenBank数据库、欧洲生物信息学研究所的EMBL数据库、以及蛋白质数据库PDB等。
在工具方面,生物信息学研究者需要掌握多种软件和编程语言。Python和R语言是生物信息学中最常用的编程语言,它们拥有丰富的生物信息学相关软件包。例如,Python的Biopython库提供了处理生物序列、解析文件格式、访问在线数据库等功能。R语言的Bioconductor项目则包含了大量用于基因组数据分析的软件包。此外,还有许多专门的生物信息学软件,如用于序列比对的BLAST、用于多序列比对的ClustalW和MUSCLE、用于系统发育分析的MEGA等。
中国在生物信息学工具和数据库建设方面也取得了显著成果。例如,中国科学院北京基因组研究所开发了BIG Data Center,整合了多种基因组学数据资源。华大基因开发了BGI Online等生物信息学分析平台,为科研人员提供便捷的数据分析服务。这些本土化的工具和数据库不仅服务于国内科研,也在国际上产生了重要影响。
序列比对算法与数据库应用
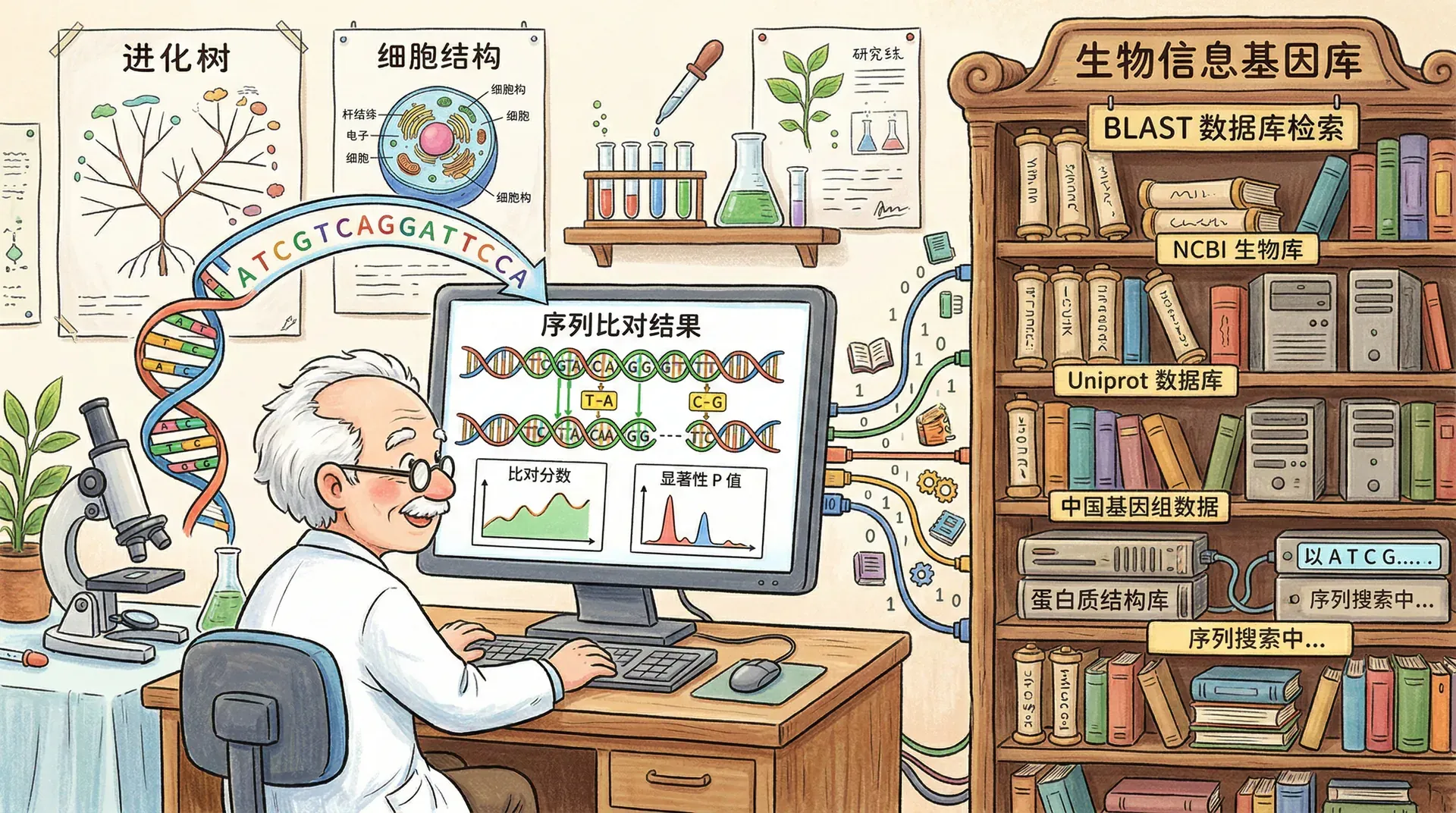
序列比对是生物信息学中最基本也是最重要的操作之一。通过比较两条或多条生物序列,我们可以推断它们之间的相似性,进而了解它们的进化关系、功能相似性以及结构特征。序列比对的核心思想是在两条序列之间找到最佳的对应关系,这种对应关系能够最大化序列之间的相似性。
序列比对可以分为两类:双序列比对和多序列比对。
双序列比对又分为全局比对和局部比对。全局比对试图在整个序列长度上找到最佳匹配,适用于长度相近且整体相似的序列。最著名的全局比对算法是Needleman-Wunsch算法,由尼德尔曼(Needleman)和温奇(Wunsch)在1970年提出。该算法采用动态规划的思想,通过构建一个二维矩阵来计算最优比对。
局部比对则是寻找序列中最为相似的局部区域,适用于部分相似或者长度差别较大的序列。最经典的局部比对算法是Smith-Waterman算法,由史密斯(Smith)和沃特曼(Waterman)在1981年提出。该算法同样使用动态规划,但允许比对从任意位置开始和结束。
在序列比对中,我们需要定义一个评分系统来衡量比对的质量。通常,匹配的碱基或氨基酸会获得正分,错配会获得负分,而引入空位(gap)也会扣分。
以下是一个简化的序列比对评分示例。假设我们比对两条短序列“ACGT”和“AGCT”,使用简单的评分规则:匹配得1分,错配扣1分,引入空位扣2分。
虽然动态规划算法能够找到最优比对,但其时间复杂度较高,对于大规模数据库搜索来说过于缓慢。为了解决这一问题,研究者开发了启发式算法。BLAST(基本局部比对搜索工具,中文全称“基本局部相似性搜索工具”)是最著名的序列比对工具,由史蒂芬·F·阿尔奇尔(Stephen F. Altschul)等人于1990年开发。BLAST通过寻找短的高度相似片段(通常称为“种子”),然后对这些种子进行扩展,从而快速找到局部比对。虽然BLAST不保证找到最优解,但其速度要快得多,能够在短时间内完成整个数据库的搜索。
下方展示了不同序列比对算法在不同序列长度下的计算时间(秒)比较:
可以看出,BLAST算法的效率明显高于传统的动态规划算法。
在实际应用中,序列比对常常需要访问各种生物序列数据库。GenBank(美国国家生物技术信息中心维护的核酸序列数据库)包含了来自超过10万个物种的数据。UniProt(通用蛋白质资源库)是蛋白质序列和功能信息的综合数据库,整合了Swiss-Prot和TrEMBL两个数据库。蛋白质数据库PDB(蛋白质结构数据库)则收录了利用X射线晶体学、核磁共振等方法解析的蛋白质和核酸的三维结构数据。
中国也建立了多个重要的生物信息学数据库。例如,中国科学院建立的中国国家基因库(CNGB),是继美国NCBI、欧洲EBI和日本DDBJ之后的第四个国家级基因库。该基因库不仅存储和管理大量的基因组数据,还提供数据分析和共享服务。此外,还有专门针对特定物种或研究领域的数据库,如水稻基因组数据库、中国人群基因组变异数据库等。
基因预测与功能注释方法
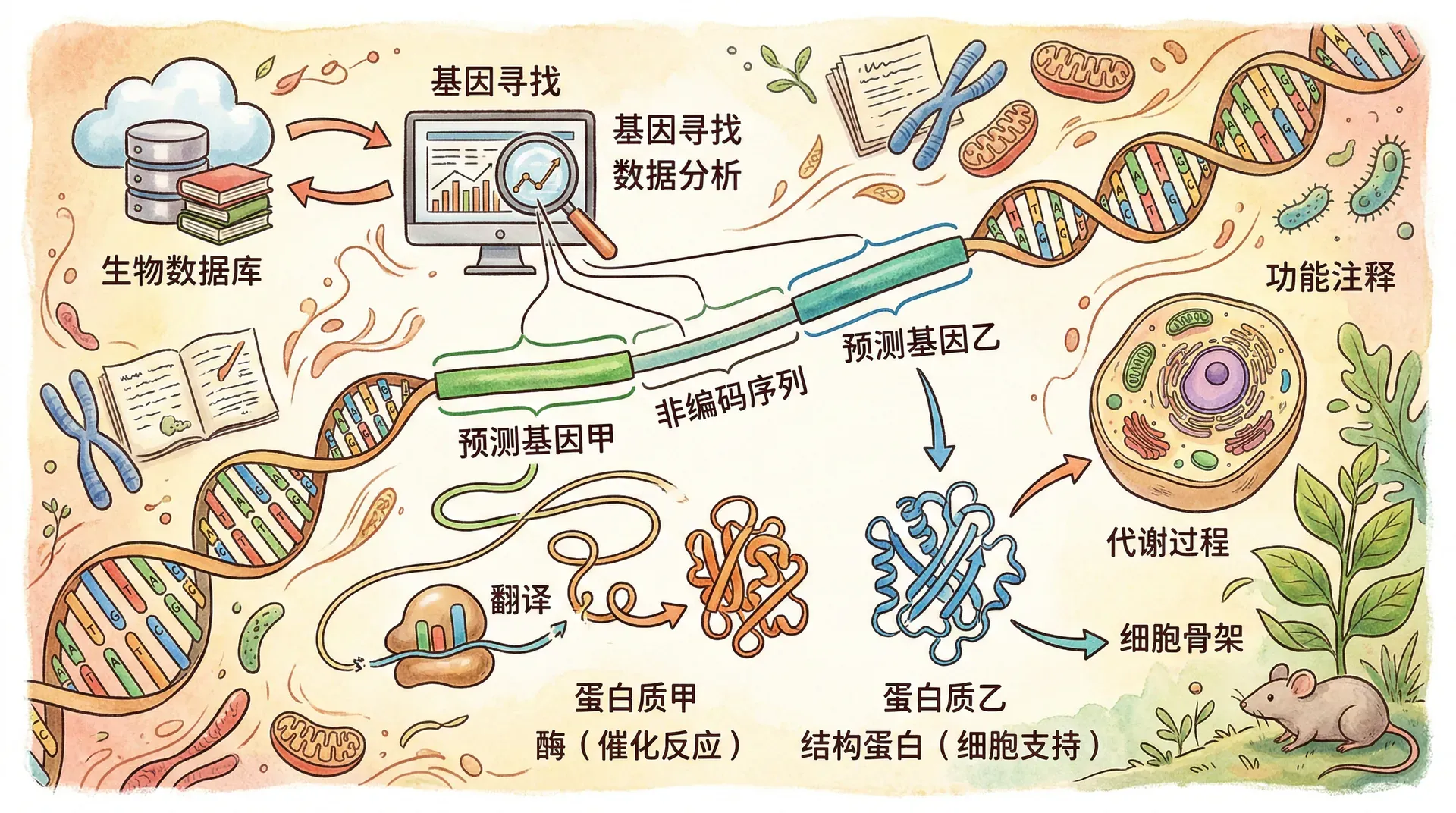
基因预测是生物信息学中的一项重要任务,其目标是在DNA序列中识别出编码蛋白质的基因。对于原核生物来说,基因预测相对简单,因为原核生物的基因通常没有内含子,基因之间的非编码区也较短。然而,真核生物的基因结构要复杂得多,包含外显子、内含子、启动子、终止子等多种元件,这使得真核生物的基因预测成为一个具有挑战性的问题。
基因预测方法可以分为三类:从头预测(ab initio)、基于同源性的预测和基于转录证据的预测。
-
从头预测方法不依赖于其他物种的序列信息,而是基于统计模型来识别基因特征。这类方法通常使用隐马尔可夫模型(HMM)或神经网络来学习基因的序列特征,如起始密码子、终止密码子、剪接位点等。常用的从头预测软件包括GeneMark、Augustus和GENSCAN等。
-
基于同源性的预测方法则利用已知基因的序列信息。通过将待预测序列与已知基因进行比对,可以推断出新序列中可能的基因位置。这种方法的准确性较高,但依赖于参考序列的质量和相似度。
-
基于转录证据的预测方法使用RNA测序(RNA-seq)数据或EST(表达序列标签)数据来直接确定基因的转录区域。由于这些数据反映了基因的实际表达情况,因此准确性很高。
在实际应用中,研究者通常会结合多种方法来提高基因预测的准确性。例如,先使用从头预测方法得到初步结果,然后用同源性信息和转录证据进行验证和修正。
以下比较了三种主要基因预测方法的特点:
基因预测完成后,下一步是进行功能注释,即推断所预测基因的生物学功能。功能注释通常通过序列相似性搜索来实现。如果一个新预测的基因与已知功能的基因有高度相似的序列,则可以推测它们具有相似的功能。常见的功能注释工具包括BLAST、InterProScan和HMMER等。
基因本体(Gene Ontology,简称GO)是功能注释中广泛使用的标准化术语体系。GO将基因功能分为三个主要类别:分子功能、生物过程和细胞组分。通过GO注释,我们可以系统地描述基因的功能特征。例如,一个基因可能被注释为“具有ATP结合活性”(分子功能)、“参与细胞周期调控”(生物过程)、“定位于细胞核”(细胞组分)。
KEGG(京都基因与基因组百科全书)是另一个重要的功能注释资源。KEGG数据库收录了大量的代谢通路、信号转导通路以及其他生物学通路的信息。通过将基因映射到KEGG通路上,可以了解基因在整个生物网络中的作用。中国科学家也在功能基因组学研究中作出了重要贡献,比如对水稻、小麦等重要农作物开展了大规模的功能基因组研究。
下图展示了不同基因预测方法的准确性比较。准确性通过敏感性(Sensitivity,预测到的真实基因比例)和特异性(Specificity,预测结果中真实基因的比例)来衡量。
蛋白质结构预测与分子对接
蛋白质的功能很大程度上取决于其三维结构。常见的实验方法如X射线晶体学和核磁共振(NMR)能够解析蛋白质结构,但这些实验方式通常成本高、耗时长。相比之下,计算机辅助预测蛋白质结构则高效且经济,因此成为生物信息学中最具挑战性也是最活跃的研究方向之一,被称为“蛋白质折叠问题”。
蛋白质结构预测的主要方法如下所示:
其中,同源建模是目前最为可靠的方法。当目标蛋白与已知结构的蛋白质序列相似度较高(一般大于30%)时,可以直接以模板结构为蓝本进行推断。此方法广泛应用于新药设计、蛋白质工程等领域。
线程法(Threading/Fold recognition)适用于序列相似性较低但可能拥有相似折叠类型的蛋白,通过在已知结构的骨架进行穿线和评分来选取最合理结构。从头预测则完全不依赖于模板,而是根据物理化学原理(如能量最小化)在理论上对任何蛋白序列进行三维结构建模,但因构象组合极其庞大,目前主要局限于较短的蛋白质。
2020年,DeepMind公司开发的AlphaFold2在蛋白质结构预测领域取得突破性进展。该系统基于深度学习模型,在CASP14国际竞赛中实现了与实验方法可比的结构预测精度。2021年,AlphaFold2的代码和全球大量蛋白质结构预测数据库对外开放,极大推动了结构生物学和药物研发进展。
近年来,中国科学家也在蛋白质结构解析和结构生物学领域取得了一系列国际领先成果。例如,清华大学颜宁团队解析了葡萄糖转运蛋白GLUT1、施一公团队构建了剪接体和核糖体等高分辨率结构。这些工作极大推动了膜蛋白、超大分子复合体的功能研究和应用。
蛋白质结构预测的一个重要实际应用是分子对接(Molecular Docking)。分子对接是一种计算方法,用于预测小分子配体(如药物分子)与蛋白质受体的结合方式和亲和力。在新药筛选中,科学家通常需要评估药物分子是否可以与特定蛋白的活性位点有效结合。现代的分子对接流程主要可以总结为:
- 活性位点识别:确定配体可能结合的区域。
- 采样与构象生成:在该区域内生成配体的多种空间排列和构象。
- 评分函数评估:评价不同结合模式的能量或亲和力,筛选最优结合方式。
常用对接软件工具包括AutoDock、DOCK、Glide等。下表为示例:
下图展示了蛋白质结构预测准确度与模板序列相似度的关系。可以看到,同源建模在高相似度条件下具有更低的平均结构偏差(RMSD),而当相似度降低时,结构预测难度显著增加。
机器学习在生物信息学中的应用
机器学习,作为人工智能的重要分支,通过从数据中学习模式用于预测和决策。近年来,受益于算力提升和算法创新,机器学习已广泛应用于生物信息学领域,包括基因预测、蛋白质结构预测和疾病诊断等,展现出强大潜力。
生物信息学中常见的机器学习方法主要有监督学习、非监督学习与深度学习:
- 监督学习:利用带标签的数据训练模型,学习输入与输出的映射。例如基因预测,通过已知的正负样本训练分类器以识别新序列中的基因。典型算法包括支持向量机(SVM)、随机森林和神经网络等。
- 非监督学习:处理无标签数据,主要用于发现隐藏模式,如聚类在基因表达谱分析中应用广泛,能发现具有类似表达的基因群,对揭示共调控或共功能基因组有重要意义。
- 深度学习:使用多层神经网络自动提取特征,能对复杂的结构和序列数据建模。卷积神经网络(CNN)常用于DNA序列调控元件分析、转录因子结合位点预测等;循环神经网络(RNN)及长短期记忆网络(LSTM)则适合处理序列数据,如蛋白质结构和剪接位点的预测。
在应用机器学习时,需防止模型过拟合(即在训练集表现优秀但泛化能力弱)。常用对策包括交叉验证、正则化,以及增加训练数据量等。
深度学习作为机器学习中的前沿技术,其端到端的建模能力在生物信息学中极具突破性。以AlphaFold2为代表,利用注意力机制和残差网络,通过学习大量已知蛋白质的结构信息,实现高精度三维结构预测。这不仅彰显了深度学习强大的特征表达能力,也为结构生物学和药物研发带来了新机遇。
目前,中国在人工智能与生物信息学的交叉领域也已投入大量资源,涌现出众多基于机器学习开发的生物信息学工具。例如,华为、腾讯、阿里等公司与各大研究机构相继推出基因组分析、疾病预测、药物研发等人工智能服务,推动相关产业和科学发展。
下表对常见机器学习方法在生物信息学中的典型应用、优势与挑战进行了整合总结:
机器学习应用仍面临数据质量(如噪声、缺失值和批次效应)、模型可解释性(特别是在医学领域)以及计算资源等挑战。然而,随着高质量数据积累、算法进步和运算能力提升,机器学习对生物信息学的推动作用必将愈发显著。
下图展示了不同机器学习模型在基因功能预测任务中的性能(准确率及F1分数)对比:
本节练习
1. 在序列比对中,Smith-Waterman算法与Needleman-Wunsch算法的主要区别是什么?
A. Smith-Waterman算法用于多序列比对,Needleman-Wunsch算法用于双序列比对
B. Smith-Waterman算法进行局部比对,Needleman-Wunsch算法进行全局比对
C. Smith-Waterman算法速度更快,但准确性较低
D. Smith-Waterman算法只能用于蛋白质序列,Needleman-Wunsch算法只能用于核酸序列
2. BLAST算法相比Smith-Waterman算法的主要优势是什么?
A. BLAST算法能找到最优解
B. BLAST算法速度快,适合大规模数据库搜索
C. BLAST算法准确性更高
D. BLAST算法可以比对蛋白质三维结构
3. 在基因预测中,基于转录证据的预测方法使用的数据是:
A. 其他物种的基因序列
B. 蛋白质结构数据
C. RNA测序或EST数据
D. 代谢通路信息
4. AlphaFold2在蛋白质结构预测中使用的核心技术是:
A. 分子动力学模拟
B. X射线晶体学
C. 深度学习
D. 同源建模
5. 在生物信息学中,Gene Ontology(GO)主要用于:
A. 序列比对
B. 基因功能注释
C. 蛋白质结构预测
D. 系统发育分析
6. 请简要说明为什么序列相似性可以用来推断生物功能和进化关系。这一原理的局限性是什么?
7. 请讨论机器学习在生物信息学中的应用前景和面临的主要挑战。