基因组学与转录组学
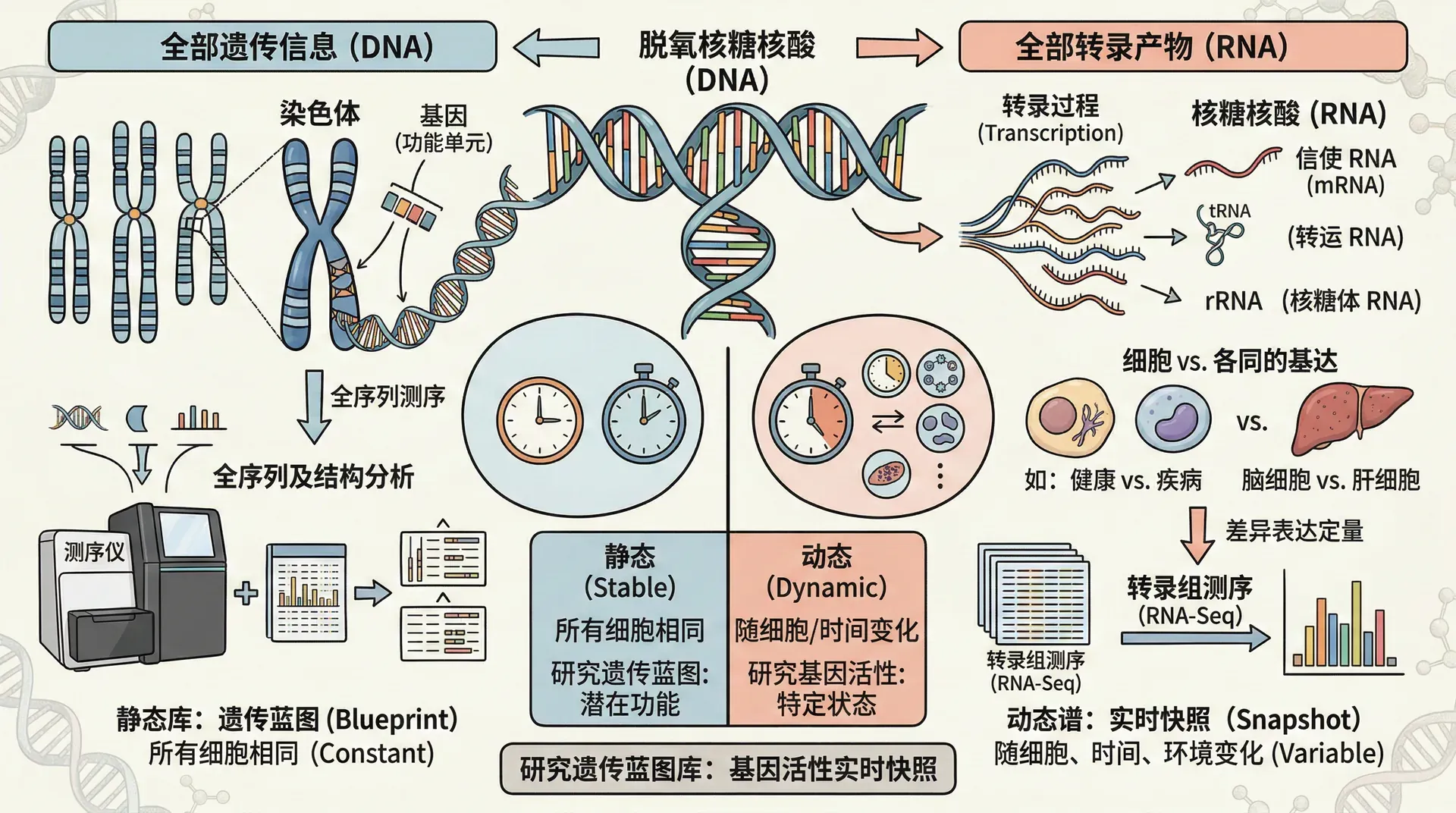
基因组学(Genomics)和转录组学(Transcriptomics)是解析生物体生命本质的两大核心研究方向。基因组学关注于生物体全部基因组DNA序列的结构、功能、进化及其相互作用,致力于揭示基因的排列顺序、遗传信息以及调控网络。随着高通量测序技术的发展,科学家能够对整个基因组进行全面分析,识别与疾病、性状、进化等相关的遗传变异。这为疾病诊断、精准医疗、作物改良等领域带来了革命性的变革。
转录组学则重点研究细胞在特定生理或病理状态下,基因被转录为RNA分子的整体表达情况(即转录组)。通过RNA测序(RNA-seq)等技术,研究者不仅可以量化每个基因的表达水平,还能发现新的转录本、可变剪接事件和非编码RNA,从而深入理解细胞功能、分化命运以及对外界刺激的响应。转录组学常用于探索疾病机制、细胞异质性、生物信息调控等科学问题。
基因组学为我们描绘了生命的“蓝图”,而转录组学则展示了生命“运行时的动态画卷”。两者密切结合,有助于揭示基因与表型、环境之间的复杂关系,为推动基础研究和生物医学应用提供坚实基础。
基因组测序技术的发展历程
基因组学作为现代生物学的核心领域,经历了从手工测序到高通量自动化测序的革命性转变。这一发展历程不仅改变了我们对生命本质的认识,也为医学、农业和生物技术的创新提供了强大的工具。
早期测序技术的奠基
20世纪70年代,两种革命性的DNA测序方法几乎同时诞生。美国科学家弗雷德里克·桑格开发了双脱氧链终止法(Sanger测序),而美国化学家马克西姆和吉尔伯特则发明了化学降解法。这两种方法标志着人类首次能够准确读取DNA序列的碱基顺序,为基因组学的诞生奠定了基础。
桑格测序法的原理类似于“在建造过程中故意制造断裂点”。通过在DNA合成过程中加入特殊的双脱氧核苷酸,使DNA链在特定位置终止延伸,最终通过分离不同长度的DNA片段来推断原始序列。这一方法在1977年首次完整测定了噬菌体φX174的基因组,全长仅5386个碱基,但这已经是人类科学史上的重大突破。
桑格测序法凭借其高准确性和可靠性,在长达30年的时间里一直是基因组测序的金标准,直到21世纪初才逐渐被新一代测序技术所取代。
自动化测序时代的到来
进入20世纪90年代,测序技术迎来了自动化革命。美国应用生物系统公司(ABI)开发的毛细管电泳测序仪,将荧光标记技术与计算机自动化分析相结合,使测序速度提升了数百倍。每个碱基用不同颜色的荧光染料标记,当DNA片段通过激光检测器时,计算机自动识别并记录碱基序列。
自动化测序技术的发展可以用数据来直观展示其进步速度。在人类基因组计划启动初期,测序成本高达每碱基10美元,而到了2003年项目完成时,成本已降至每碱基0.01美元。
新一代测序技术的突破
2005年,454公司推出了第一台商业化的新一代测序仪,标志着基因组学进入了高通量测序时代。与传统桑格测序相比,新一代测序技术具有并行处理的优势,能够同时对数百万个DNA片段进行测序。这就像从“单车道”升级为“多车道高速公路”,极大地提高了通行效率。
新一代测序技术主要包括三类具有代表性的技术平台。
-
第一类以因美纳(Illumina)公司的可逆终止子技术为代表,通过桥式扩增和合成测序方法,实现了超高通量的短片段(短读长)测序。
-
第二类平台包括安捷伦(Ion Torrent)的半导体测序技术,利用DNA合成过程中释放的氢离子进行碱基检测。
-
第三类为单分子实时测序技术,以太平洋生物(PacBio)和牛津纳米孔(Oxford Nanopore)公司为代表,可以直接读取长达数万甚至数十万碱基的DNA片段,有效突破了传统短读长的限制。
第三代测序技术的单分子长读长特性,使其在解析复杂基因组结构、检测结构变异和表观遗传修饰方面具有独特优势,特别适合中国科学家在研究复杂农作物基因组时使用。
人类基因组计划与中国贡献
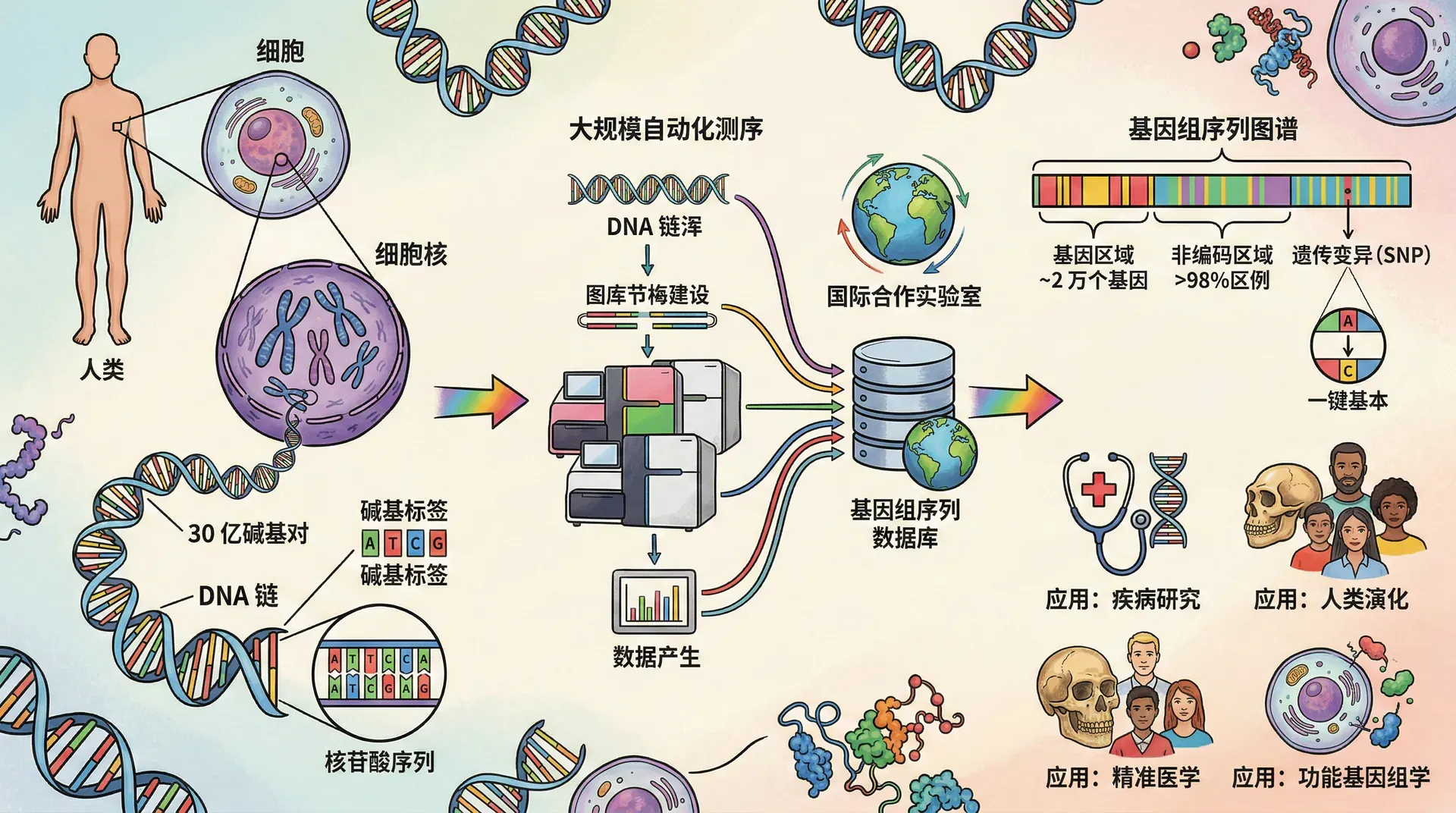
人类基因组计划(Human Genome Project,HGP)是人类科学史上最宏大的国际合作项目之一,其影响力可与曼哈顿计划和阿波罗登月计划相媲美。这个项目不仅绘制了人类基因组的完整图谱,也深刻改变了生物学研究的范式。
人类基因组计划的启动与实施
1990年,由美国、英国、日本、法国、德国和中国等多国科学家共同参与的人类基因组计划正式启动。项目的主要目标是测定人类基因组中全部30亿个碱基对的序列,识别所有基因,并将这些信息存储在数据库中供全世界科学家免费使用。
项目实施过程中采用了“分层策略”。首先将人类基因组切割成大约15万个细菌人工染色体(BAC)克隆,每个克隆包含约15万个碱基对。然后对每个克隆进行测序,最后通过计算机将这些片段拼接成完整的基因组序列。这种策略类似于完成一幅巨大的拼图游戏,需要精确的组织和协调。
人类基因组计划的进展情况如下信息所示:
中国在人类基因组计划中的角色
中国于1999年9月正式加入人类基因组计划,成为继美、英、日、德、法之后的第六个参与国。尽管加入时间较晚,但中国科学家承担了人类3号染色体短臂上约3000万个碱基对的测序任务,约占人类基因组总量的1%。这一贡献使中国成为唯一参与该计划的发展中国家,展现了中国在基因组学领域的实力。
中国人类基因组计划的实施主要由中国科学院遗传研究所人类基因组中心(北京)和华大基因研究中心(深圳)承担。特别值得一提的是,华大基因在项目后期发挥了重要作用,不仅按时完成了分配的测序任务,还建立了完善的基因组测序平台和生物信息学分析体系。
中国在人类基因组计划中的参与,不仅提升了国际科学界对中国科研实力的认可,更重要的是培养了一批优秀的基因组学人才,为中国后续在基因组学领域的快速发展奠定了坚实基础。
中国基因组学研究的后续发展
人类基因组计划完成后,中国在基因组学领域的研究持续推进并取得了多项重要成果。2008年,深圳华大基因完成了第一个亚洲人(炎黄一号)基因组测序,这是首个由中国科学家独立完成的个人基因组。随后,华大基因又主导完成了大熊猫基因组、家蚕基因组等多个重要物种的基因组测序项目。
在农业基因组学方面,中国科学家取得了令世界瞩目的成就。2002年,中国科学院和国家杂交水稻工程技术研究中心领衔完成了籼稻基因组精细图谱。2010年,深圳华大基因联合多国科学家完成了黄瓜、西瓜等重要蔬菜作物的基因组测序。这些成果不仅具有重要的科学价值,也为中国农业育种提供了重要的基因资源。
比较基因组学与进化基因组学
比较基因组学和进化基因组学是基因组学领域的两个重要分支,它们通过比较不同物种或同一物种不同个体的基因组序列,揭示生命的进化历程和物种间的亲缘关系。这种研究方法就像考古学家通过比较不同时期的文物来推断历史演变一样,通过比较基因组序列来追溯生命的历史。
比较基因组学的基本原理
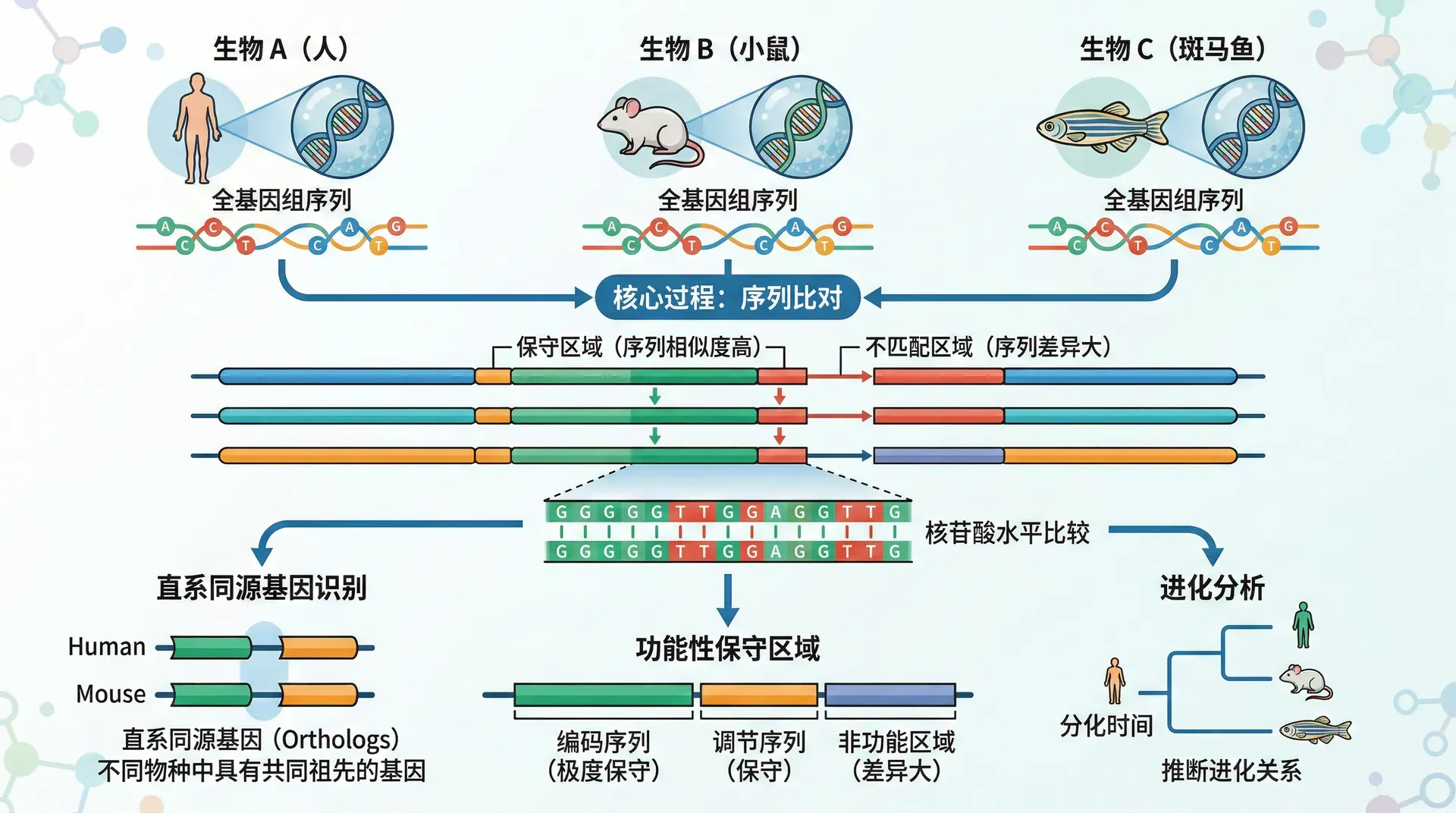
比较基因组学的核心思想是通过比对不同物种的基因组序列,识别功能保守区和变异区。保守区往往承担着基础且重要的生物学功能,在长期进化过程中因选择压力而保持稳定;而变异区则反映了物种为适应不同环境所产生的遗传创新。
下表以数据举例,展示人类与几类典型生物的基因组相似性,可以直观感受进化关系及保守区与变异区的对比:
尽管有高低不同的相似度,比较基因组学研究还揭示了一个现象:基因组大小与物种复杂度并不完全相关。例如:
- 人类基因组约30亿个碱基对、约20,000个蛋白质编码基因;
- 水稻基因组仅约4亿个碱基对,但蛋白质编码基因数量却超过37,000个。
这说明决定生物复杂性的并非基因数量本身,而是基因调控和功能的多样性。
进化基因组学的研究方法
进化基因组学致力于利用大量基因组数据,重建物种的进化历程和系统发育关系。科学家通常会比对不同物种同源基因间的序列差异,例如下列表格简述主流程方法:
分子钟假说认为:在无强烈选择压的基因序列区段,中性突变会以相对稳定的速率积累。因此,两个物种间序列差异的数量可以用来估算它们分化的年代,这个过程好像用“序列的不同”去读懂“物种分离的时间”。
在科学研究实践中,中国学者贡献突出:在2014年,昆明动物研究所联合团队完成树鼩基因组测序,为灵长类进化研究提供参考模型;另外,2016年,北京大学团队揭示了藏族人群的高原遗传适应机制,发现EPAS1和EGLN1基因变异帮助藏族人适应低氧。
进化基因组学的深入研究不仅有助于揭示生命的起源与演化,还能为疾病基因解析、模型构建等领域提供重要理论和数据支持。许多人类疾病相关基因都能在模式生物中找到高度保守的同源基因,通过这种比较可以深化我们对疾病分子机制的理解。
基因组重排与物种分化
基因组重排是物种进化过程中的重要机制之一。染色体的倒位、易位、重复和缺失等结构变异,可以导致基因功能的改变或新基因的产生,从而推动物种分化。比较基因组学研究发现,人类和黑猩猩之间存在多处染色体重排事件,其中人类2号染色体是由两条祖先染色体融合而成的。
在植物基因组进化中,全基因组复制(Whole Genome Duplication,WGD)事件扮演着重要角色。研究表明,被子植物在进化过程中至少经历了两次全基因组复制事件,这为植物提供了大量的遗传原料,促进了新性状的产生和物种多样性的增加。中国科学家对棉花基因组的研究发现,棉花在约600万年前经历了全基因组复制事件,这与棉花纤维发育相关基因的扩增密切相关。
下方总结了几种重要模式生物的基因组特征和进化关系:
转录组测序技术与数据分析
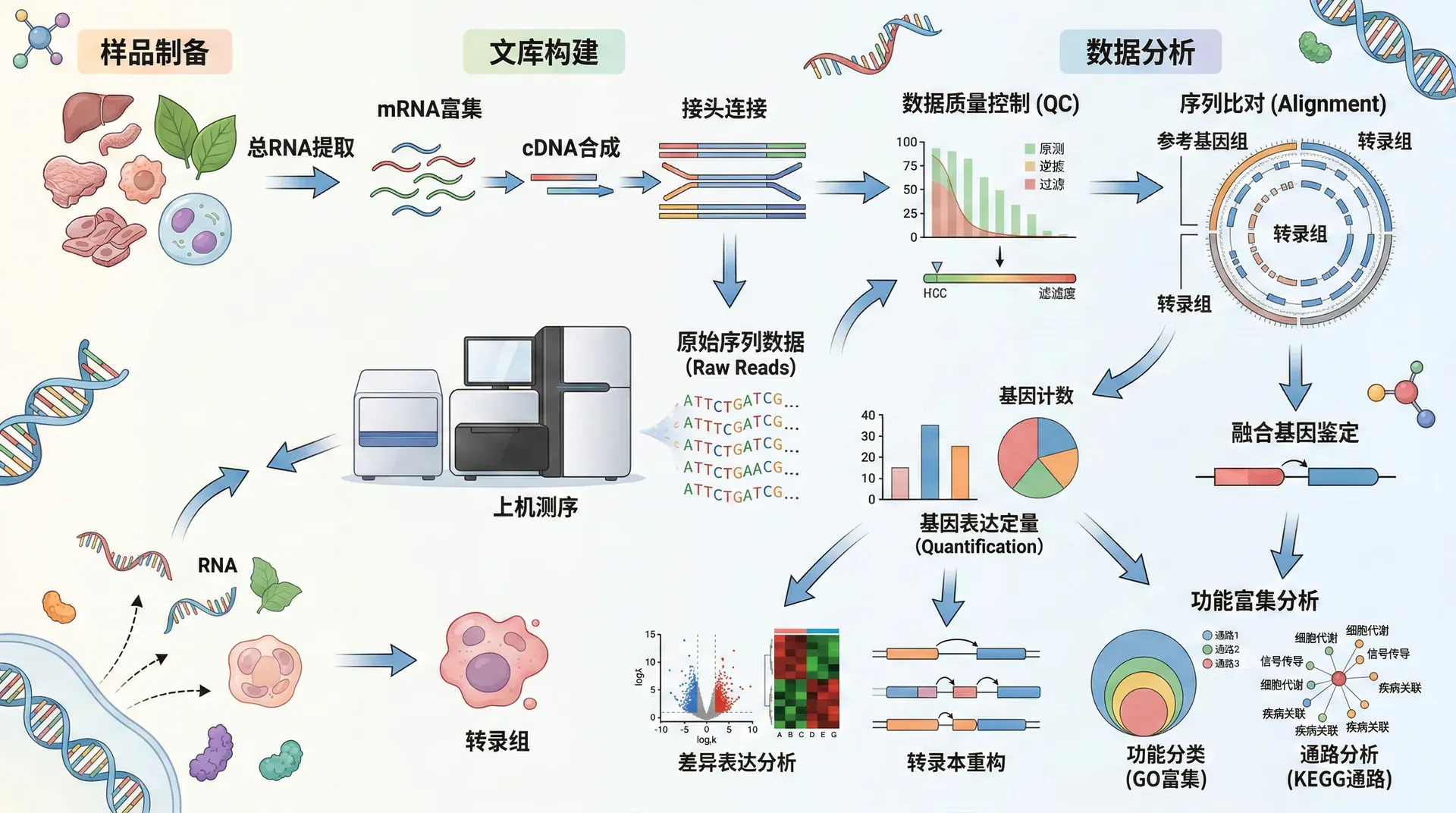
转录组是指某一特定细胞、组织或生物体在特定生理状态下转录出的所有RNA分子的集合。转录组学研究通过分析基因的表达模式,揭示基因如何响应内外环境变化,调控生物体的生长发育和生理功能。
转录组测序技术的演进
早期的转录组研究主要依赖于基因芯片技术。基因芯片通过在固体基片上固定数千个已知基因的探针,利用荧光标记的样本RNA与探针杂交,检测不同基因的表达水平。这种技术虽然可以同时检测大量基因的表达,但受限于预先设计的探针,无法发现新的转录本,且动态范围有限。
RNA测序技术(RNA-seq)的出现彻底改变了转录组研究的格局。2008年,基于新一代测序技术的RNA-seq方法正式应用于转录组研究。该技术首先将样本中的RNA反转录成cDNA,然后对cDNA进行高通量测序,最后通过生物信息学分析将测序读段比对到参考基因组,定量各个基因的表达水平。
RNA-seq技术具有多方面的优势。
-
它不依赖于预先设计的探针,可以检测所有转录本,包括新的转录本和未知基因。
-
它的动态范围极广,可以准确检测从极低到极高表达水平的基因。
-
它能够精确到单个碱基的分辨率,可以检测可变剪接、RNA编辑等转录后修饰事件。
转录组数据的生物信息学分析
转录组测序产生的原始数据需要经过一系列生物信息学分析步骤才能获得有意义的生物学结论。整个分析流程可以分为数据质控、序列比对、表达量定量和差异表达分析等关键步骤。
数据质控是第一步,主要目的是评估测序数据的质量并去除低质量序列。常用的质控工具如FastQC(测序质量控制),可以评估碱基质量分布、GC含量、序列重复率等指标。如果发现低质量碱基或接头序列污染,需要使用Trimmomatic(去除低质量序列及接头)等工具进行修剪和过滤。
序列比对是将测序读段定位到参考基因组或转录组的过程。常用的比对软件包括HISAT2(高速比对工具)、STAR(剪接读段比对软件)等。这些软件能够处理RNA-Seq特有的剪接比对问题,准确识别跨越外显子边界的读段。比对完成后,使用SAMtools(结果整理与索引工具)等对比对结果进行排序和索引,为后续分析做准备。
表达量定量是计算每个基因或转录本表达水平的步骤。常用的定量方法包括基于比对的计数(如featureCounts)和基于模型的估算(如kallisto、Salmon)。表达量通常用FPKM(每千个碱基的转录本每百万映射读取片段数)或TPM(每百万转录本)来表示,这些标准化方法可以消除基因长度和测序深度的影响,使不同样本和不同基因之间的表达量具有可比性。
差异表达分析是转录组研究的核心内容,通过比较不同实验条件下基因表达的差异,识别响应特定处理或参与特定生物学过程的关键基因。常用的统计方法包括DESeq2和edgeR,它们基于负二项分布模型来检验基因表达的显著性差异。
转录组学在中国的应用研究
中国科学家在转录组学研究领域取得了多项重要成果,如下:
单细胞测序技术的原理与应用
单细胞测序技术作为基因组学研究的前沿工具,可以在单个细胞层面解析基因表达和遗传特征,揭示组织、器官中的细胞复杂性和异质性。这一技术的进步,让我们能够从“群体画像”进入到“个体特写”,大大提升了研究的分辨率。
单细胞测序的技术原理
单细胞测序最大的难点是如何从极少量的单细胞物质中获取足够可用于测序的遗传信息。以一个典型真核细胞为例,RNA含量仅约10皮克(10^-11克),远少于常规测序的需求。为此,科学家们发展了多种细胞分离与核酸扩增策略。
主要单细胞分离方法对比如下:
单细胞测序技术经历了三个关键演进阶段:
- 2009年首次实现单细胞RNA测序,开创领域;
- 2014年液滴微流控大幅提升通量并促成商业化;
- 2020年多组学联合测序平台(如scATAC-seq+scRNA-seq)推动更多生物学应用。
单细胞转录组数据分析
单细胞数据与传统转录组学最大差异在于其超大规模(动辄数万~数十万细胞)、高噪声(如dropout现象)及显著的细胞异质性,因此其分析流程也需针对性优化。常见流程如下:
单细胞测序在组织类型识别中的应用
单细胞测序的前沿应用
单细胞技术正推动多个前沿领域重大突破。例如:
发育生物学:单细胞追踪细胞命运,重建胚胎细胞谱系图,揭示从受精卵到组织器官每一阶段的分化轨迹。例如,中国团队用单细胞技术重构了小鼠胚胎发育“细胞地图”。
肿瘤异质性研究:单细胞测序揭示肿瘤内部不同亚群,为靶向治疗和耐药机制研究打开新思路。北京大学在肝癌单细胞研究中,发现多种具有高转移和耐药能力的肿瘤亚群。
神经科学:通过单细胞分析,科学家已识别出100多种大脑神经元类型,美中联合团队绘制了小鼠脑的全细胞类型图谱。
空间转录组(Spatial transcriptomics)是单细胞领域的新前沿,通过空间定位结合高通量测序,能在保留组织切片空间信息的同时揭示单细胞甚至亚细胞分辨率的表达情况。例如,科学家可用这一技术在原位追踪肿瘤与微环境的相互作用。
中国在单细胞测序领域的贡献
中国科学家在单细胞测序创新和应用上取得了国际瞩目的成果:
本节练习
1. 假设一个研究项目需要测序10个人类全基因组(每个基因组30亿碱基对),分别计算在1995年、2005年和2020年完成该项目所需的成本。基于成本数据,分析测序技术进步对基因组学研究普及化的影响。
2. 已知人类与黑猩猩的基因组序列相似度为98.8%,人类基因组大小约为30亿碱基对。请计算人类和黑猩猩之间存在多少个碱基差异。结合进化理论,解释为什么这1.2%的差异能够导致两个物种如此显著的表型差异。
3. 下表显示了某植物在干旱胁迫处理前后5个基因的表达量(FPKM值)。请识别哪些基因可能参与干旱响应,并说明判断依据。
4. 某研究团队希望研究肝脏再生过程中细胞类型的变化和相互作用。请设计一个单细胞测序实验方案,包括样本采集时间点、细胞分离方法、测序平台选择和关键分析步骤。
5. 针对以下三个研究目标,请选择最合适的基因组学技术并说明理由:
(A)鉴定一种新发现的水稻品种中可能与高产性状相关的基因
(B)研究乳腺癌肿瘤组织中不同癌细胞亚群的基因表达差异
(C)分析人群中与2型糖尿病易感性相关的遗传变异